 2021, Vol. 35
2021, Vol. 35



芳香腈类化合物是重要的有机合成中间体, 可以作为酰胺、胺、酮、羧酸、酯的合成原料, 因而在生物医药、农药、香料等领域均有广泛应用[1]. 腈类合成方法有卤代烃的氰化[2]、酰胺和醛肟的脱水[3-4]、Sandmeyer反应[5]、C—H键的氰化等[6-7], 但上述方法均涉及多步反应或使用了剧毒氰化物[8-9], 不符合绿色化学发展理念, 因此开发绿色高效的腈类合成方法具有重要意义.
2009年, Oishi T等[10]制备了非均相Ru(OH)x/Al2O3催化体系, 可在一定条件下催化氨化氧化芳香醇到芳香腈, 但使用了高成本的贵金属;2013年, Dornan等[11]制备了均相Copper/TEMPO催化体系, 催化氨化氧化芳香醛到芳香腈, 虽使用了非贵金属钴催化反应, 但催化剂不易分离; 近年来报道的Co-N-C催化剂具有稳定性好且催化活性高的优点, 在液相催化、电催化、光催化等领域均有广泛应用[12-16]. 2014, Jagadeesh等[17]所报道的非均相非贵金属Co-N-C体系, 实现了高效催化氧化醇到醛. 在上述工作的基础上, 我们以Co(NO3)2·6H2O和1-丁基-3-甲基咪唑氢溴酸盐为前驱体, Vulcan XC72R为载体, 采用浸渍法制备负载型Co-N-C催化剂, 对催化氨氧化α-芳香醇连续合成α-芳香腈进行了探索研究, 为α-芳香腈的绿色合成提供了基础实验数据.
1 实验部分 1.1 仪器和试剂XRD测试使用Bruke D8 Advance射线衍射仪进行, 测试条件为: Cu Kα射线源, Ni滤光片, 管电压40 kV, 管电流40 mA, 广角扫描范围10°~90°; 表面元素种类及元素状态通过美国Thermo ESCALAB 250XI型X射线光电子能谱仪进行分析; HRTEM测试使用美国FEI TecnaiG2F20型透射电子显微镜, 能谱oxford x-max 80T. 日本岛津公司气相色谱仪和气质联用仪(气质联用仪条件:GSBP-5型毛细管柱(30 m×0.32 mm×0.25 μm)、载气为氦气. 所用试剂六水合硝酸钴Co(NO3)2·6H2O、1-丁基-3-甲基咪唑氢溴酸盐(BmimBr)、苯甲醇、对甲基苯甲醇、3-苯基-2-烯丙醇、2, 6-二氯苯甲醇、胡椒醇、2-环己基甲醇、正辛醇、叔戊醇、正十六烷、乙醇、乙酸乙酯均购买于阿拉丁试剂有限公司(市售分析纯), 使用前未做纯化处理, Vulcan XC72R型碳粉购买于赛博电化学材料有限公司.
1.2 催化剂的制备准确称取Co(NO3)2·6H2O 1.83 g、1-丁基-3-甲基咪唑氢溴酸盐(BmimBr) 4.38 g于烧杯中, 室温搅拌10 min, 待烧杯内固体完全溶解, 溶液呈深蓝色, 加入Vulcan XC72R型碳粉2 g、无水乙醇40 mL, 持续搅拌12 h后旋干, 80 ℃置于烘箱中干燥12 h, 研磨充分、放入管式炉, 在N2 氛围下以10 ℃/min的升温速率分别升高到400、500、600、700、800、900 ℃, 保持2 h. 将不同温度焙烧的Co-N-C催化剂, 记为Co-N-C/T(T为焙烧温度). 将Co-N-C/700催化剂浸入0.5 mol/L的H2SO4溶液中, 80 ℃烘箱中存放6 h后过滤, 去离子水洗涤3次, 记为Co-N-C/700(H+).
1.3 催化剂活性评价针对不同底物, 在优化后的条件下, 分别取相应的α-芳香醇2.5 mmol于聚四氟乙烯反应釜内衬中, 加入催化剂0.2 g、氨水1 mL、叔戊醇20 mL后密封反应釜, 充入O2 0.5 MPa, 于120 ℃反应20 h后冷却至室温过滤. 打开反应釜, 真空抽滤后用50 mL乙酸乙酯稀释滤液, 加入0.1 g内标物正十六烷. 采用GCMS进行定性分析, 设置进样口温度为250 ℃, 柱箱温度从80 ℃以15 ℃/min的速度升温至280 ℃, 离子源温度为220 ℃, 接口温度为220 ℃; 采用GC进行定量分析, 采用内标法分别计算不同醇的转化率和相应腈的选择性.
2 结果与讨论 2.1 不同焙烧温度催化剂的催化氨氧化性能以对甲基苯甲醇的催化氨氧化为考察对象, 不同焙烧温度得到的催化剂催化氨氧化一锅合成对甲基苯甲腈的实验结果如表 1所示. 从表 1可知. 不同焙烧温度得到的催化剂都有一定的催化氨氧化活性, 随着焙烧温度的增加, 对甲基苯甲醇的转化率和产物对甲基苯甲腈的选择性均有提高的趋势, 其中催化剂Co-N-C/700对对甲基苯甲醇的转化率和产物对甲基苯甲腈的选择性均达到100%.
| 表 1 不同催化剂的转化率和选择性 Table 1 The convertion and selectivity of different catalysts |
使用国际衍射数据中心(ICDD)的WinXpow(Stoe)软件和粉末衍射文件(PDF)数据库完成粉末图案的处理和分配. 不同焙烧温度(400、500、600、700、800和900 ℃)得到的催化剂的XRD图谱分别标记为(a)、(b)、(c)、(d)、(e)、(f), 700 ℃焙烧后酸洗处理后的催化剂XRD图谱标记为(g), 如图 1所示. 由图 1可知, 在2θ= 25°处均存在一个宽峰, 归属于石墨C结构的(002)平面衍射峰; (a)、(b)仅具有碳载体峰的石墨烯结构, 没有金属Co特征峰, 表明钴以高度分散或无定形存在; 随着热解温度的升高, (c)、(d)、(e)、(f)都在2θ= 44.1°, 51.4°和75.9°处均显示3个峰, 这些峰接近于金属Co的面心立方结构的(111), (200)和(220), 晶面(PDF#15-0806), 从中还可以看出随着热解温度的提高金属钴的峰强度也显著增加, XRD测试结果证实了催化剂结构中金属钴的存在, 晶体Co的出现可能是在高温下含Co前体被还原, 且随着温度的升高Co元素更易于还原.

|
图 1 不同催化剂的XRD谱图 Fig.1 XRD patterns of different catalysts a. Co-N-C/400; b. Co-N-C/500; c. Co-N-C/600; d. Co-N-C/700; e. Co-N-C/800; f. Co-N-C/900; g. Co-N-C/700(H+) |
结合表 1的实验结果, 初步表明晶体Co的存在可能是提高底物的转化率和产物的选择性的影响因素. 在Co-N-C/400和Co-N-C/500催化的反应中, 对甲基苯甲醇的转化率仅为28%. 随着还原温度的升高, 催化剂结构中晶体Co的衍射峰强度增加, 对甲基苯甲醇的转化率不断提高, 催化剂Co-N-C/700对应的原料转化率和产物选择性都达到100%. 随着还原温度的进一步提高, 结晶度最高的Co-N-C/900催化反应中, 对甲基苯甲醇的转化率仅为63.7%, 推断Co的结晶度不是决定催化活性的主要因素. 为了验证这一推论, 将催化剂Co-N-C/700用酸处理, 得到Co-N-C/700(H+), XRD光谱如图 1(g), 图中仅具有较弱的Co光谱峰, 这表明酸洗后Co的结晶度显著下降, 对应的催化剂应用于对甲基苯甲醇的催化氨氧化, 其转化率依然保持在78%. 为了深入研究Co的结晶度对催化活性的影响, 对催化剂Co-N-C/700(H+)进行了SAED和HRTEM测试分析, 结果如图 2所示. 从图 2可知, 其局部区域的SAED图案仅显示石墨烯碳层的衍射环, 而没有涉及钴晶粒的衍射点, HRTEM图中仅有石墨烯碳层结构, 没有发现显著的钴晶粒结构. 这与前述的XRD测试图 1(g)相一致. 也进一步证实Co的结晶度不是决定催化活性的主要因素.

|
图 2 Co-N-C/700(H+)样品的(SAED)图案(a)和HRTEM图像(b) Fig.2 (SAED) patterns (a) and HRTEM image (b) of Co-N-C/700(H+) samples |
近年来, 低共熔溶剂(DES)被人们广泛应用于催化材料的制备过程中, 这类催化材料除了具有非均相催化剂易分离、可回收、比表面积大的优点, 还兼具均相催化剂转化率高、选择性好的优点[18-19]. 实验通过混合无机金属盐(Co(NO3)2·6H2O)和季铵盐型离子液体(1-丁基-3-甲基咪唑氢溴酸盐)得到均相的低共熔溶剂前驱体, 这种低共熔体系可同时将金属原子以及含N有机配体构建在前驱体中, 再进一步将含Co-N配体的前驱体固定在碳载体表面上, 经不同温度焙烧, 得到系列金属负载型催化剂.
为进一步探究不同焙烧温度催化剂的催化性能差异, 对XRD表征中有晶体Co出现的Co-N-C进行N 1s的XPS高分辨谱图分析, 结果如图 3所示.

|
图 3 不同催化剂的N 1s高分辨谱图 Fig.3 XPS spectra in N 1s region for different catalysts (a) Co-N-C/600; (b) Co-N-C/700; (c) Co-N-C/800; (d) Co-N-C/900 |
由图 3可知, N 1s的XPS谱图中有3个峰, 分别归属于吡啶N(398.6 eV)、Co-N中心(399.3 eV)和石墨N(401 eV)的特征峰[20]. 随着焙烧温度的升高, 吡啶N的含量逐渐降低, 石墨N的含量逐渐升高, Co-N中心的含量先升高后降低, 可认为Co-N中心的存在是吡啶N向石墨N转化的中间态. 结合表 1分析, Co-N-C/700中, Co-N中心的含量最高, 体现出最佳的催化活性; 随着Co-N中心含量的降低, 反应的催化活性随之降低, 认为Co-N中心的存在, 在催化反应中起着关键作用[21-22]. 对比XRD表征分析和催化性能评价结果, 酸洗后的Co物种可能以原子或者高度分散的形式存在, 导致催化活性明显下降, 表明晶体Co的出现在催化反应过程中并没有起主导作用; 同时, 随着焙烧温度升高, 催化剂的催化活性出现下降趋势, 这可能是随着温度的升高, 一方面有机N的氧化流失增加, 另一方面催化剂的团聚也相对增加, 都会导致Co-N中心活性位降低, 进而影响催化活性. 综合分析认为, Co-N中心的存在对整个反应的顺利进行起到了决定性的作用, 而DES中有效的Co-N配位作用也有助于Co-N中心的形成和保持.
2.4 催化剂对不同底物的催化氨氧化性能固定Co-N-C/700为催化剂, 对含不同取代基的α-芳香醇和脂肪醇的催化氨氧化反应制备对应有机腈的实验结果如表 2所示.
| 表 2 Co-N-C/700对不同反应底物的催化氨氧化性能 Table 2 Catalytic ammoxidation performance of different reactants from Co-N-C/700 |

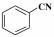


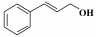


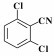
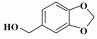

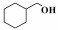

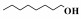

在催化苯甲醇氨化氧化制备苯甲腈反应的基础上, 将Co-N-C/700进一步应用于其他α-芳香醇和部分脂肪醇的氨化氧化反应中. 结果表明, 该催化剂对其它α-芳香醇的氨氧化合成腈的反应也表现出了较好的催化活性, 主产物为相对应的α-芳香腈; 当反应底物为非α-羟基醇时, 得到的对应产物腈的选择性很低, 这里可能有两个方面的原因:一是在催化氧化阶段α-羟基易于被氧化成对应的中间产物; 二是α-羟基醇对应的催化氨氧化产物α-腈类存在着π-π共轭结构, 具有较高的热力学稳定性.
2.5 催化剂的稳定性实验以苯甲醇的催化氨氧化制备苯甲腈为考察对象, 在相同反应条件下对Co-N-C/700催化剂重复使用性能进行测试, 结果如图 4所示.

|
图 4 Co-N-C/700的循环利用 Fig.4 The reusability of Co-N-C/700 |
将首次反应混合物过滤后得到的催化剂, 置于乙醇溶液中, 超声10 min后离心, 再用乙醇洗涤离心两次, 于80 ℃烘箱干燥24 h后直接投入下一次使用. 从图 4可知, 在使用1、2、3、4和5次后, 苯甲腈的收率分别为100%、96%、96%、93%和90%, 表明Co-N-C/700催化剂具有较好的稳定性.
3 结论以Co(NO3)2·6H2O和1-丁基-3-甲基咪唑氢溴酸盐为Co-N前驱体, 通过浸渍法负载在Vulcan XC72R碳粉上, 在不同温度下焙烧制备了系列负载型Co-N-C催化剂, 并应用于α-芳香醇和部分脂肪醇连续催化氨氧化反应直接合成对应的有机腈. 在优化的实验条件下, Co-N-C/700催化剂对催化α-芳香醇氨氧化直接制备相应的α-芳香腈具有优良的催化性能, 相关α-芳香醇的转化率和对应α-芳香腈的选择性可达100%, 且稳定性良好. 该工作为有机腈的高效绿色合成提供了基础研究数据.
| [1] |
Fleming F F, Yao Li-hua, Ravikumar P C, et al. Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore[J]. J Med Chem, 2010, 53(22): 7902–7917.
DOI:10.1021/jm100762r |
| [2] |
Khemnar A B, Bhanage B M. Copper catalyzed nitrile synthesis from aryl halides using formamide as a nitrile source[J]. RSC Adv, 2014, 4(26): 13405–13408.
DOI:10.1039/C3RA48075E |
| [3] |
Kim H S, Kim S H, Kim J N. Highly efficient Pd-catalyzed synthesis ofnitriles from aldoximes[J]. Tetrahed Lett, 2009, 50(15): 1717–1719.
DOI:10.1016/j.tetlet.2009.01.150 |
| [4] |
Jiang Nan, Ragauskas A J. Ultrasound-promoted synthesis of nitriles from aldoximes under ambient conditions[J]. Tetrahed Lett, 2010, 51(34): 4479–4481.
DOI:10.1016/j.tetlet.2010.06.079 |
| [5] |
Sandmeyer T. Ueber die ersetzung der amid-gruppe durch chlor, brom und cyan in den aromatischen substanzen[J]. Berichte Der Deutschen Chemischen Gesellschaft, 1884, 17(2): 2650–2653.
DOI:10.1002/cber.188401702202 |
| [6] |
Anbarasan P, Schareina T, Beller M. Recent developments and perspectives in palladium-catalyzed cyanation of aryl halides: Synthesis of benzonitriles[J]. Chem Soc Rev, 2011, 40(10): 5049–5067.
DOI:10.1039/c1cs15004a |
| [7] |
Li Jie, Ackermann L. Cobalt-catalyzed CH cyanation of arenes and heteroarenes[J]. Angew Chem Int Ed, 2015, 127(12): 3706–3709.
DOI:10.1002/ange.201409247 |
| [8] |
Zhang Jun-li, Chen Xiao-rong, Hu Tong-jie, et al. Highly efficient Pd-catalyzed cyanation of aryl chlorides and arenesulfonates with potassium ferrocyanide in aqueous media[J]. Catal Lett, 2010, 139(1/2): 56–60.
DOI:10.1007/s10562-010-0385-1 |
| [9] |
Schareina T, Zapf A, Cotté A, et al. A versatile protocol for copper-catalyzed cyanation of aryl and heteroaryl bromides with acetone cyanohydrin[J]. Adv Syn Catal, 2011, 353(5): 777–780.
DOI:10.1002/adsc.201000200 |
| [10] |
Oishi T, Yamaguchi K, Mizuno N. Catalytic oxidative synthesis of nitriles directly from primary alcohols and ammonia[J]. Angew Chem Int Ed, 2009, 48(34): 6286–6288.
DOI:10.1002/anie.200900418 |
| [11] |
Dornan L M, Cao Qun, Flanagan J C A, et al. Copper/TEMPO catalysed synthesis of nitriles from aldehydes or alcohols using aqueous ammonia and with air as the oxidant[J]. Chem Commun, 2013, 49(54): 6030.
DOI:10.1039/c3cc42231c |
| [12] |
Wu J C S, Lin Zhi-an, Tsai F M, et al. Low-temperature complete oxidation of BTX on Pt/activated carbon catalysts[J]. Catal Today, 2000, 63(2/4): 419–426.
|
| [13] |
Zhu Nai-shu, Ma Shi-ning, Sun Xiao-feng. Nitrogen-doped carbon fiber paper by active screen plasma nitriding and its microwave heating properties[J]. ACS Appl Mater Interf, 2016, 8(51): 35606–35613.
DOI:10.1021/acsami.6b10262 |
| [14] |
Wang Ting, Shi Shao-jun, Li Yu-hong, et al. Study of microstructure change of carbon nanofibers as binder-free anode for high-performance lithium-ion batteries[J]. ACS Appl Mater Interf, 2016, 8(48): 33091–33101.
DOI:10.1021/acsami.6b11996 |
| [15] |
Share K, Cohn A P, Carter R, et al. Role of nitrogen-dopedgraphene for improved high-capacity potassium ion battery anodes[J]. ACS Nano, 2016, 10(10): 9738–9744.
DOI:10.1021/acsnano.6b05998 |
| [16] |
Carraro F, Calvillo L, Cattelan M, et al. Fast one-pot synthesis of MoS2/crumpled graphene p-n nanonjunctions for enhanced photoelectrochemical hydrogen production[J]. ACS Appl Mater Interf, 2015, 7(46): 25685–25692.
DOI:10.1021/acsami.5b06668 |
| [17] |
Jagadeesh R V, Junge H, Beller M. Green synthesis of nitriles using non-noble metal oxides-based nanocatalysts[J]. Nat Commun, 2014, 5: 4123.
DOI:10.1038/ncomms5123 |
| [18] |
Zhang Qing-hua, De Oliveira Vigier K, Royer S, et al. Deep eutectic solvents: Syntheses, properties and applications[J]. Chem Soc Rev, 2012, 41(21): 7108–7146.
DOI:10.1039/c2cs35178a |
| [19] |
Qureshi Z S, Deshmukh K M, Bhanage B M. Applications of ionic liquids in organic synthesis and catalysis[J]. Clean Tech Environ Policy, 2014, 16(8): 1487–1513.
DOI:10.1007/s10098-013-0660-0 |
| [20] |
Duan Ya-nan, Song Tao, Dong Xiao-su, et al. Enhanced catalytic performance of cobalt nanoparticles coated with a N, P-codoped carbon shell derived from biomass for transfer hydrogenation of functionalized nitroarenes[J]. Green Chem, 2018, 20(12): 2821–2828.
DOI:10.1039/C8GC00619A |
| [21] |
Niu Ke-xing, Yang Bao-ping, Cui Jin-feng, et al. Graphene-based non-noble-metal Co/N/C catalyst for oxygen reduction reaction in alkaline solution[J]. J Power Sour, 2013, 243: 65–71.
DOI:10.1016/j.jpowsour.2013.06.007 |
| [22] |
Noyori R, Yamakawa M, Hashiguchi S. Metal-ligand bifunctional catalysis: A nonclassical mechanism for asymmetric hydrogen transfer between alcohols and carbonyl compounds[J]. J Org Chem, 2001, 66(24): 7931–7944.
DOI:10.1021/jo010721w |