 2022, Vol. 36
2022, Vol. 36



2. 山东临沂生态环境监测中心, 山东 临沂 276001;
3. 临沂大学, 山东 临沂 276005
2. Linyi Eco-Environment Monitoring Center, Linyi 276001, China;
3. Linyi University, Linyi 276005, China
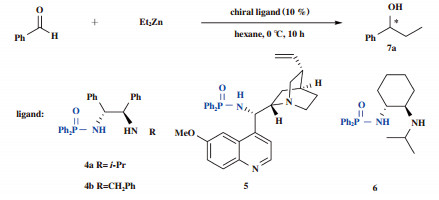
手性是自然界最重要的属性之一[1]. 绝大多数的药物是由手性化合物组成的, 拥有药理作用的药物一般都是手性专一的. 许多具有重要生理作用的药物分子及其中间体都有α-芳基丙醇结构片段, 如具有抗病毒活性的α-乙基-1-金刚烷甲醇[2-4]; 治疗神经系统疾病的药物托莫西汀的医药中间体[5-6]; 用于预防支气管哮喘发作的麻黄碱[7-9]以及对用于合成复杂化合物的亚甲基环丙烷化合物[10-13]等都含有α-芳基丙醇结构框架. 人工合成α-芳基丙醇类化合物的需求越来越大, 因此也促进了α-芳基丙醇不对称合成的发展.
众所周知, 手性α-芳基丙醇化合物合成方法有很多种, 例如加氢还原合成法[14-15], 格式试剂加成合成法[16-17]、手性试剂拆分合成法[18-19]以及不对称催化合成方法[20], 其中氢化还原方法及格式试剂法因反应活性较高, 很难做到高手性选择性, 手性拆分方法虽然能得到较高光学纯的产物, 但也有其局限性, 比如收率只有50%, 造成大量的物料浪费, 不符合目前所推崇的绿色化学理念; 目前最推崇、最热门的合成方法是不对称催化法, 因为手性试剂能够提供唯一的手性环境, 因此可以高收率高ee值地合成目标化合物.

|
图 1 含有α-芳基丙醇构架的化合物举例 Fig.1 Compounds containing α-aryl propanol structures |
使用烷基金属试剂催化合成二级醇的报道有很多, 常用金属试剂为锌试剂、钛试剂、锂试剂等[21-22], 相比其他金属试剂, 锌试剂因其低价、低毒备受关注. 因5价膦化合物化学性质稳定, 毒性较低, 手性磷酰胺类化合物在不对称催化合成方向被广泛使用[23-26], 为了进一步扩宽其应用范围, 下面我们介绍了一种使用手性磷酰胺类化合物4a不对称催化合成α-芳基丙醇化合物的方法.
1 实验部分 1.1 试剂与仪器R, R-1, 2-二苯基乙二胺购于化夏化学北京公司; 一水合对甲苯磺酸、邻苯二甲酸酐及二乙基锌均购于上海易恩化学技术有限公司; 一水合肼购于上海泰坦科技股份有限公司; 其他试剂均购于烟台市双双化工有限公司.
气相色谱仪(型号:9790Ⅱ, 浙江福立仪器公司); 核磁共振波谱仪(型号:Bruker AVANCE Ⅲ 400MHz型, 瑞士Bruker公司); 高分辨质谱仪(型号:Impact Ⅱ UHR-TOF型, 瑞士Bruker公司); 高效液相色谱仪(型号:LC-20ATvp型, 日本岛津).
核磁谱图1H NMR(400 MHz)和13C NMR(100 MHz)是以CDCl3氘代DMSO等为溶剂进行测试, 氢谱以δ=7.26、碳谱以δ=77.0作为化学位移的基准; 核磁氢谱中的单重峰、双重峰、三重峰、四重峰、混合峰分别用字母s、d、t、q、m表示; 实验中所使用的试剂可商业获得; 旋光度由旋光仪测得, 其表示方法为(c 10 mg/mL, 溶剂); GC数据由气相色谱仪及手性色谱柱测得(检测器类型:FID; 手性硅胶毛细管柱(Chirasil Dex CB column, 25 m Length×0.32 mm× 0.25 mm film thickness)); HPLC数据由高效液相色谱仪及手性色谱柱测得.
1.2 实验方法 1.2.1 手性磷酰胺类化合物4a的合成方法(图 2)

|
图 2 手性磷酰胺类化合物4a合成路线 Fig.2 Synthesis route of chiralphosphoramide 4a |
反应瓶中依次加入甲苯20 mL, R, R-1, 2-二苯基乙二胺(20 g 94.2 mmol)、邻苯二甲酸酐(14.0 g 94.2 mmol)以及一水合对甲苯磺酸(16.2 g 94.2 mmol), 反应液加热回流反应6 h, 使用薄层色谱法进行中控, 直至无原料剩余后进行下一步操作. 冷却至室温后进行减压过滤得滤饼, 滤饼用饱和碳酸钠溶液溶解后使用二氯甲烷萃取得有机相, 有机相使用无水硫酸钠搅拌干燥. 干燥结束抽滤得到抽滤液, 抽滤液使用旋转蒸发仪浓缩至干燥的固体物, 固体物不再进行其他纯化即为氨基保护的R, R-1, 2-二苯基乙二胺(化合物1).
将上述化合物1、二氯甲烷、三乙胺(11.4 g 113.0 mmol)加入反应瓶, 控温0~5 ℃搅拌条件下, 将使用DCM稀释后的二苯基膦酰氯(22.3 g 94.2 mmol)缓慢加入反应瓶中, 加入完毕将反应体系升温至室温搅拌反应2 h后每隔30 min使用薄层色谱法监控反应情况, 直至无化合物1剩余后进行下一步操作. 反应液加入冰水混合物中进行淬灭并使用二氯甲烷进行萃取得有机相, 有机相使用无水硫酸钠搅拌干燥30 min, 搅拌结束抽滤得到抽滤液, 抽滤液使用旋转蒸发仪浓缩干燥, 对所得的浓缩物使用柱层析方法进行分离提纯(VC4H8O2∶VC6H14=1∶3),得化合物2(44.0 g, 产率86%). m.p. 134~135 ℃; [α]D25.1 78.5(c 1.00, CH2Cl2); 1H NMR(400 MHz, CDCl3)δ 4.43(t, J=10.2 Hz, 1H), 5.41(q, J=20.2 Hz, 1H), 5.81(d, J=9.2 Hz, 1H), 7.04-7.23(m, 10H), 7.26-7.47(m, 8H), 7.59-7.72(m, 4H), 7.73-7.78(m, 2H); 13C NMR(100 MHz, CDCl3)δ 56.7, 61.3, 123.2, 127.1, 127.2, 127.7, 127.0, 128.0, 128.1, 128.2, 128.3, 129.1, 131.4, 131.5, 131.6, 131.7, 131.8, 131.9, 132.2, 132.4, 132.5, 133.1, 133.7, 136.4, 140.8, 168.6. HRMS(ESI): (m/z)calcd. for C34H28N2O3P [M+H]+: 543.1759, found: 543.1766.
三口反应瓶中加入无水乙醇100 mL、化合物2(44.0 g, 81.0 mmol)、水合肼(81 mL)后回流搅拌反应120 min. 使用薄层色谱法进行中控, 直至无化合物2剩余后进行下一步操作至反应结束. 将反应瓶放置在常温水浴中进行降温, 当温度达到30 ℃时, 使用旋转蒸发仪对反应液进行减压浓缩得浓缩物. 所得浓缩物加入无水乙醇及无水乙醚, 室温条件下搅拌萃取30 min后进行加压抽滤, 向所得滤液中加入无水硫酸钠搅拌干燥30 min. 抽滤得滤液, 滤液浓缩至干燥后进行柱层析纯化(VC4H8O2∶VC6H14=1∶3), 得化合物3(32.4 g, 产率97%). m.p. 215~216 ℃; s[α]D27.2 76.9(c 1.00, CH2Cl2); 1H NMR(400 MHz, CDCl3)δ 4.45(t, J=10.2 Hz, 1H), 5.41(q, J=20.4 Hz, 1H), 5.82(d, J=9.2 Hz, 1H), 7.03-7.24(m, 10H), 7.23-7.45(m, 8H), 7.56-7.73(m, 4H), 7.75-7.81(m, 2H); 13C NMR(100 MHz, CDCl3)δ 61.18, 61.41, 126.72, 127.13, 127.21, 127.73, 128.14, 128.11, 128.16, 128.21, 128.23, 131.31, 131.33, 131.48, 131.52, 132.01, 132.08, 132.11, 132.24, 132.46, 132.64, 133.68, 141.54, 141.52, 142.4; HRMS(ESI): (m/z)calcd. for C26H26N2OP [M+H]+: 413.1704, found: 413.1902.
向反应瓶中依次加入化合物3(31.2 g, 78.0 mmol)、乙腈、碳酸钾(21.6 g, 156.0 mmol)及碘代异丙烷(26.5 g, 156.0 mmol), 将反应瓶放入油浴中加热搅拌至回流, 搅拌反应2 h后无需降温, 直接将反应液使用旋转蒸发仪进行减压浓缩得浓缩物, 浓缩物加入纯化水搅拌溶解无机盐, 有机物使用二氯甲烷萃取并使用无水硫酸钠干燥经过柱层析提纯得化合物4a(28.7 g收率81%). m.p. 204~207 ℃; [α]D21.0 18.5(c 1.00, CH2Cl2); 1H NMR(400 MHz, CDCl3)δ 0.89(d, J=6.2 Hz, 3H), 0.97(d, J=6.4 Hz, 3H), 2.51-2.65(m, 1H), 4.00(d, J=6.0 Hz, 1H), 4.29-4.37(m, 1H), 4.45-4.65(m, 1H), 7.05-7.16(m, 5H), 7.17-7.22(m, 2H), 7.23-7.33(m, 7H), 7.34-7.44(m, 2H), 7.51-7.61(m, 2H), 7.65-7.75(m, 2H); 13C NMR(100 MHz, CDCl3)δ 21.6, 24.1, 45.3, 60.9, 66.0(d, J=6.8 Hz), 126.9, 127.1, 127.3, 127.7, 127.9, 128.0, 128.04, 128.1, 128.3, 131.24, 131.28, 131.36, 131.4, 131.7, 131.8, 132.1, 132.2, 133.1, 133.14, 140.67, 140.69, 140.8; 31P NMR(162 MHz, CDCl3)δ 22.4;. HRMS(ESI): (m/z)calcd. for C29H32N2OP [M+H]+: 455.2174, found: 455.2183.
1.2.2 其他手性磷酰胺类化合物的合成手性磷酰胺类化合物4b的合成如下:4b的合成方法与4a的几乎一致, 仅仅是把碘代异丙烷更换为碘代苯甲烷, 不再进行赘述.
手性磷酰胺类化合物5的合成如下:5的合成方法详见文献[27].
手性磷酰胺类化合物6的合成如下:6的合成方法详见文献[26].
1.2.3 化合物7的表征数据(S)-(-)-1-苯基-1-丙醇(7a):收率:91%; 无色液体; 94% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 7.5 min(S), tR = 7.9 min(R)]; 1H NMR(400 MHz, CDCl3: δ 7.41-7.22(m, 5H), 4.60(t, J = 6.4 Hz, 1H), 1.90(s, 1H), 1.87-1.68(m, 2H), 0.92(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 144.8, 128.6, 127.6, 126.1, 76.2, 32.0, 10.3.
(R)-(-)-1-(2-甲基苯基)-1-丙醇(7b):收率:95%; 无色液体; 87% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 16.3 min(S), tR = 19.3 min(R)]; 1H NMR(400 MHz, CDCl3: δ 7.46(d, J = 7.6 Hz, 1H), 7.28-7.08(m, 3H), 4.87(t, J = 6.4 Hz, 1H), 2.34(s, 3H), 1.81-1.69(m, 3H), 0.98(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 142.9, 134.8, 130.5, 127.3, 126.4, 125.4, 72.3, 31.1, 19.2, 10.5.
(R)-(-)-1-(3-甲基苯基)-1-丙醇(7c):收率:90%; 无色液体; 82% 的ee值是通过手性GC检测获得[柱温:140 ℃, tR = 30.88 min(S), tR = 31.72 min(R)]; 1H NMR(400 MHz, CDCl3: δ 7.62-6.67(m, 1H), 7.19-6.53(m, 3H), 4.94-3.99(m, 1H), 2.36(s, 3H), 1.92-1.63(m, 3H), 0.92(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 141.8, 137.3, 129.2, 126.1, 76.1, 32.0, 21.2, 10.3.
(R)-(-)-1-(4-甲基苯基)-1-丙醇(7d):收率:92%; 无色液体; 84% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 12.74 min(R), tR = 17.89 min(S)]; 1H NMR(400 MHz, CDCl3: δ 7.24-7.22(m, 2H), 7.17-7.15(m, 2H), 4.56(m, J = 6.8Hz, 1H), 2.35(s, 3H), 1.92-1.64(m, 3H), 0.91(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 144.7, 128.5, 126.8, 123.2, 76.2, 32.0, 21.6, 10.3.
(S)-(-)-1-(2-氯苯基)-1-丙醇(7e):收率:89%; 无色液体; 83% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 9.98 min(S), tR = 11.10 min(R)]; 1H NMR(400 MHz, CDCl3: δ 7.50-7.57(m, 1H), 7.35-7.25(m, 2H), 7.19(m, 1H), 5.50-4.62(m, 1H), 1.99(m, 1H), 1.90-1.66(m, 2H), 0.99(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 142.1, 132.2, 129.5, 128.5, 127.3, 127.2, 72.2, 30.6, 10.2.
(S)-(-)-1-(3-氯苯基)-1-丙醇(7f):收率:96%; 无色液体; 76% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 16.13 min(S), tR = 21.43 min(R)]; 1H NMR(400 MHz, CDCl3: δ 7.35-7.33(m, 1H), 7.30-7.16(m, 3H), 4.60-4.55(m, 1H), 1.98(s, 1H), 1.84-1.67(m, 2H), 0.91(t, J = 7.4, 3H); 13C NMR(100 MHz, CDCl3): δ 146.8, 134.5, 129.8, 127.7, 126.3, 124.3, 75.5, 32.1, 10.1.
(S)-(-)-1-(4-氯苯基)-1-丙醇(7g):收率:97%; 无色液体; 78% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 11.98 min(S), tR = 14.82 min(R)]; 1H NMR(400 MHz, CDCl3: δ d 7.32-7.25(m, 4H), 4.58(t, J = 6.4 Hz, 1H), 1.93(s, 1H), 1.84-1.66(m, 2H), 0.90(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 143.2, 133.3, 128.7, 127.5, 75.4, 32.1, 10.1.
(S)-(-)-1-(2-氟苯基)-1-丙醇(7h):收率:88%; 无色液体; 93% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 17.77 min(R), tR = 18.39 min(S)]; 1H NMR(400 MHz, CDCl3: δ 7.43(m, 1H), 7.25-7.19(m, 1H), 7.13-7.09(m, 1H), 7.01-6.96(m, 1H), 4.89(t, J = 6.8 Hz, 1H), 2.48(s, 1H), 1.81-1.72(m, 2H), 0.91(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 159.9(d, J = 245.1 Hz), 131.6(d, J = 13.0 Hz), 128.8(d, J = 8.1 Hz), 127.4(d, J = 4.6 Hz), 124.3(d, J = 3.1 Hz), 115.3(d, J = 22.0 Hz), 69.6, 31.0, 10.0.
(S)-(-)-1-(3-氟苯基)-1-丙醇(7i):收率:84%; 无色液体; 92% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 6.99 min(R), tR= 7.32 min(S)]; 1H NMR(400 MHz, CDCl3: δ 0.90(t, J = 7.4 Hz, 3H), 1.65-1.85(m, 2H), 2.71(m, 1H), 4.55(t, J = 6.6 Hz, 1H), 6.91-7.01(m, 1H), 7.01-7.11(m, 2H), 7.22-7.34(m, 1H); 13C NMR(100 MHz, CDCl3): δ 162.9(d, J = 245.8 Hz), 147.4(d, J = 6.6 Hz), 129. 8(d, J = 8.2 Hz), 121.6(d, J = 2.8 Hz), 114.2(d, J = 21.2 Hz), 112.8(d, J = 21.4 Hz), 75.2(d, J = 1.6 Hz), 31.8, 9.9.
(S)-(-)-1-(4-氟苯基)-1-丙醇(7j): 收率: 83%; 无色液体; 90% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 17.9 min(R), tR = 18.5 min(S)]; 1H NMR(400 MHz, CDCl3: δ 0.87(t, J = 7.6 Hz, 3H), 1.55-1.85(m, 2H), 2.80(s, 1H), 4.51(t, J = 6.6 Hz, 1H), 6.91-7.06(m, 2H), 7.15-7.34(m, 2H); 13C NMR(100 MHz, CDCl3): δ 162.1(d, J = 243 Hz), 140.3(d, J = 2.7 Hz), 127.6(d, J = 8.1 Hz), 115.1(d, J = 21.0 Hz), 75.2, 31.9, 10.0.
(R)-(-)-2-萘基-1-丙醇(7k): 收率: 97%; 无色液体; 87% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 10.34 min(R), tR = 12.76 min(S)]; 1H NMR(400 MHz, CDCl3: δ 7.89-7.80(m, 3H), 7.78(m, 1H), 7.54-7.42(m, 3H), 4.77(t, J = 6.6 Hz, 1H), 1.99(s, 1H), 1.96-1.79(m, 2H), 0.95(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 142.1, 133.5, 133.2, 128.4, 128.1, 127.8, 126.3, 125.9, 124.9, 124.3, 76.3, 32.0, 10.3.
(R)-(-)-1-萘基-1-丙醇(7l): 收率: 92%; 无色液体; 89% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 17.9 min(R), tR = 18.5 min(S)]; 1H NMR(400 MHz, CDCl3: δ 8.12(d, J = 7.8 Hz, 1H), 7.88(m, 1H), 7.78(d, J = 8.0 Hz, 1H), 7.64(d, J = 6.8 Hz, 1H), 7.60-7.41(m, 3H), 5.41(t, J = 5.8 Hz, 1H), 2.11-1.83(m, 3H), 1.04(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 140.4, 134.0, 130.7, 129.1, 128.1, 126.1, 125.7, 125.6, 123.4, 123.1, 72.8, 31.3, 10.7.
(S)-(+)-2-苯基-1-丙醇(7m): 收率: 84%; 无色液体; 82% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 17.9 min(S), tR = 18.5 min(R)]; 1H NMR(400 MHz, CDCl3: δ 7.48-7.45(m, 2H), 7.39-7.35(m, 2H), 7.29-7.25(m, 1H), 1.93-1.82(m, 2H), 1.81(s, 1H), 1.58(s, 3H), 0.83(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 147.9, 128.2, 126.6, 125.0, 75.1, 36.8, 29.7, 8.4.
(R)-2-(邻甲苯基)丁烷-2-醇(7n):收率:88%; 无色液体; 84% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 16.1 min(R), tR = 17.7 min(S)]; 1H NMR(400 MHz, CDCl3: δ 7.52-7.43(m, 1H), 7.53-7.32(m, 3H), 2.61(s, 3H), 2.03-1.94(m, 2H), 1.71(s, 3H), 0.90(t, J = 7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 144.6, 135.4, 132.4, 126.7, 126.7, 125.3, 76.0, 34.4, 28.7, 22.2, 8.4.
(R)-2-(间甲苯基)丁醇(7o)收率:79%; 无色液体; 91% 的ee值是通过手性GC检测获得[柱温:135 ℃, tR = 95.3 min(R), tR = 102.0 min(S)]; 1H NMR(400 MHz, CDCl3: δ 67.25-7.21(m, 3H), 7.06-7.04(m, 1H), 2.36(s, 3H), 1.82(m, J=7.4 Hz, 2H), 1.71(s, 1H), 1.53(s, 3H), 0.80(t, J=7.4 Hz, 3H); 13C NMR(100 MHz, CDCl3): δ 147.6, 137.5, 127.9, 127.1, 125.5, 121.8, 74.9, 36.6, 29.7, 21.7, 8.4.
(S)-2-(对氯苯基)丁醇(7p):收率:89%; 无色液体; 80% 的ee值是通过手性HPLC检测获得[柱温:40 ℃, tR = 20.78 min(S), tR = 21.85 min(R)]; 1H NMR(400 MHz, CDCl3: δ 0.79(t, J = 7.4 Hz, 3H), 1.53(s, 3H), 1.69(s, 1H), 1.77-1.83(m, 2H), 7.28-7.38(m, 4H); 13C NMR(100 MHz, CDCl3): δ 146.4, 132.3, 128.2, 126.6, 74.7, 36.7, 29.6, 8.3.
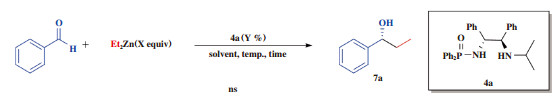
2 结果与讨论 2.1 最佳手性磷酰胺类化合物的筛选为了达到最佳的催化效果, 及在保证得到高产率的同时, 使所得化合物的ee值也较高, 我们对一系列的磷酰胺类化合物进行了选择, 测试结果如表 1, 所使用手性磷酰胺类化合物均为常见的磷酰胺类化合物, 其中手性磷酰胺类化合物4的起始物料为R, R-1, 2-二苯基乙二胺, 5的起始物料为奎尼定, 6则是由R, R-1, 2-环己基二胺合成.
| 表 1 手性磷酰胺类化合物的筛选a Table 1 Screening of chiral phosphoramide compounds |
我们使用苯甲醛和二乙基锌作为起始物料, 使用10%(摩尔分数)的手性磷酰胺类化合物, 反应温度0 ℃, 反应时间10 h, 实验结果见表 1. 数据表明, 使用手性磷酰胺类化合物4b催化不对称反应时, 7a的ee值仅有30%(表 1中entry 2); 使用手性磷酰胺类化合物5催化不对称反应时7a的收率和ee值均低于30%(表 1中entry 3); 当催化剂为手性磷酰胺类化合物6时, 目标产物7a的ee值虽然较高, 为53%, 但收率却只有66%, 达不到预期目标(表 1中entry 4); 当4a作为手性磷酰胺类化合物催化反应时, 可保证4a的收率在94%的同时, ee值也可提高至69%(表 1中entry 1).
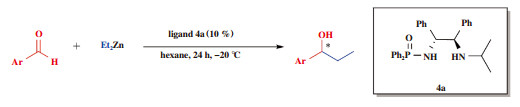
2.2 最佳反应条件的筛选我们对最佳反应条件进行了筛选, 分别就起始物料二乙基锌及催化剂4a的使用量、不同的反应溶剂、催化反应的反应时间及反应温度进行了相关优化, 实验结果见表 2. 首先我们对手性磷酰胺类化合物的使用量进行了筛选, 实验数据见表 2中entries 1-4, 如表所示, 随着起始物料二乙基锌的使用量不断升高, 目标产物7a的收率也逐渐升高, 但不幸的是, 7a的ee值却呈现出抛物线式的先增后减情况, 当底物二乙基锌使用量为2.0 eq时, 收率虽然稍低于3.0 eq的收率, 但所得7a的ee值远高于乙基锌使用量为3.0 eq时. 在研究了二乙基锌的使用量后, 我们又对手性磷酰胺类化合物的使用量进行了相应的优化, 实验数据见表 2中entries 3, 5-6, 数据表明当手性磷酰胺类化合物使用量为10%时, 所得7a的收率和ee值均与20%时相当, 考虑到手性磷酰胺类化合物使用的经济性, 优选10%的使用量. 我们对溶剂也进行了优化, 依次更换反应体系为甲苯、二氯甲烷和正己烷, 实验结果见表 2中entries 3, 7-8, 当使用正己烷作为反应溶剂时, 所得产物7a的收率及ee值均为最高值, 因此将反应溶剂定为正己烷. 此后, 我们对反应时间进行了优化, 实验数据见表 2中entries 8-10, 数据表明当反应24 h时所得7a收率与5 h、10 h相当, 但ee值为最高值, 因此将反应时间定为24 h. 最佳催化反应温度研究实验数据见表 2中entries 10-12, 我们可以清楚地从数据中看到, 温度的不断降低可有效的提高目标产物7a的ee值, 但不幸的是, 温度的降低会带来收率的不断下降. 通过对收率和ee值综合考虑, 我们将最佳温度定为了-20 ℃. 综上所述, 我们可以得到最佳的催化反应条件如下: 起始物料二乙基锌使用2 eq, 催化剂4a的使用量为10%, 正己烷为反应溶剂, 反应时间和温度分别为24 h和-20 ℃. 为了验证回收4a的催化能力, 我们使用回收所得4a作为催化剂催化反应, 催化结果见表 2中entry 13, 数据表明, 回收所得4a和新制备的4a拥有一样的催化效果.
| 表 2 反应条件的优化a Table 2 Optimization of the reaction |
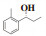
为了研究我们选择的最佳催化条件是否可以应用于各类型的反应底物, 我们对不同的反应底物进行了相关实验研究, 实验结果列与表 3. 当芳香醛苯环的邻间对位分别被甲基、氟、氯取代时, 所得α-芳基丙醇类化合物的收率均在80%以上, ee值也均在80%以上(表 3, entries 2-10); 我们尝试以萘甲醛的衍生物作为底物, 实验结果表明, 所得α-萘基丙醇类化合物的ee值均可保持在80%以上, 同时收率也可保持在90%以上(表 3, entries 11-12), 当将醛更换为酮时, 反应也可以保持较高的收率及ee值(表 3, entries 13-16).
| 表 3 底物的拓展a Table 3 Scope of the catalytic reactiona |


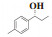

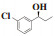
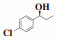

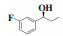
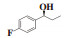
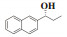
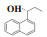
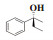
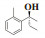
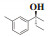
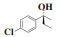
最后, 依据路易斯酸碱理论推断出一个较为合理的反应机理. 反应过程中形成的分子间四元、六元过渡态及官能团的空间位阻作用致使所得α-芳基丙醇类化合物有较高的手性. 其中, 四元过渡态是由乙基锌中的锌原子、乙基, 芳香醛中的C原子、O原子形成的; 六元环是由芳香醛中的O原子、乙基锌中的Zn原子、4a中的一个N原子、P原子、O原子以及另外一个分子的Zn形成. 此时当芳香醛中芳香基团与手性磷酰胺类化合物4a中的异丙基在同侧时, 因分子间空间位阻作用较大导致其不稳定(图 3, 中间态2); 当芳香醛中氢与手性磷酰胺类化合物4a中的异丙基在同侧时, 因氢原子较小, 与异丙基间分子间空间位阻较小, 过渡态更稳定, 因此更倾向于形成中间态1(图 3, 中间态1). 正是因为分子间形成的四元过渡态、六元过渡态及空间位阻作用导致所得α-芳基丙醇类化合物有更好的立体选择性.

|
图 3 反应机理推测 Fig.3 Proposed mechanism of the reaction |
我们使用手性磷酰胺类化合物4a为催化剂, 高选择性、高产率地催化合成手性α-芳基丙醇类化合物, 手性磷酰胺类化合物4a的普适性比较高, 合成路线简单, 起始物料廉价易得, 手性磷酰胺类化合物的使用量较为适中且可通过柱层析方法[流动相: EA/PE, V∶V=1∶8]进行回收纯化再利用, 回收所得催化剂与新制备的拥有一样的催化效果. 最后, 我们推测出一种最佳的反应机理, 认为之所以能够得到高收率和高手性专一的α-芳基丙醇类化合物的原因是催化反应过程中形成的含有四元、六元过渡态以及空间位阻作用中间态.
| [1] |
Einhorn A. Ueber derivate der orthonitrozimmtsäure[J]. Berichte der Deutschen Chemischen Gesellschaft, 1983, 16(2): 2208–2216.
|
| [2] |
a. Vícha Robert, Potáček Milan. Influence of catalytic system composition on formation of adamantane cont-aining ketones[J]. Tetrahedron, 2005, 61(1): 83-88. b. Zhang An-lin(张安林), Xu Hu(许慧), Pan Chun-yue(潘春跃), et al. Highly enantioselective asymmetric epoxidation of olefins catalyzed by chiral polyethers(手性聚醚催化烯烃的高对映选择性环氧化反应研究)[J]. J Mol Catal(China)(分子催化), 2020, 34(6): 514-524. |
| [3] |
Klimochkin Y N, Leonova M V, Korzhev I R, et al. Antiviral activity of adamantane series hydroxy deriva-tives[J]. Pharm Chem J, 1992, 26(7): 616–618.
|
| [4] |
Tafeenko V A, Prozorovskii A E, Rybakov V B, et al. Crystal and molecular structure of N-[1-(1-adamantyl)propyl]acetamide[J]. J Struct Chem, 1987, 28(1): 158–163.
DOI:10.1007/BF00749571 |
| [5] |
Rej R K, Das T, Hazra S, et al. Chemoenzymatic asymm-etric synthesis of fluoxetine, atomoxetine, nisoxetine, and duloxetine[J]. Tetrahedron: Asymmetry, 2013, 24(15/16): 913–918.
|
| [6] |
Trost B M, Fraisse P L, Ball Z T. A stereospecific ruthe-nium‐catalyzed allylic alkylation[J]. Angew Chem Int Ed, 2002, 41(6): 1059–1061.
DOI:10.1002/1521-3773(20020315)41:6<1059::AID-ANIE1059>3.0.CO;2-5 |
| [7] |
Tang X Y, Shi M. HOTf‐catalyzed rearrangement of methylenecyclopropane aryl and alkyl alcohols[J]. Eur J Org Chem, 2010, 21: 4106–4110.
|
| [8] |
Hu B, Xing S, Wang Z. Lewis acid catalyzed ring-opening intramolecular friedel-crafts alkylation of methylenecyclopropane 1, 1-diesters[J]. Org lett, 2008, 10(23): 5481–5484.
DOI:10.1021/ol802301e |
| [9] |
Lautens M, Han W, Liu J H C. MgI2-Mediated ring expansions of secondary methylenecyclopropyl amides[J]. J Am Chem Soc, 2003, 125(14): 4028–4029.
DOI:10.1021/ja028967l |
| [10] |
Postigo A, Kopsov S, Ferreri C, et al. Radical reactions in aqueous medium using (Me3Si)3SiH[J]. Org Lett, 2007, 9(25): 5159–5162.
DOI:10.1021/ol7020803 |
| [11] |
Ting R, Lermer L, Perrin D M. Triggering DNAzymes with light: A photoactive C8 thioether-linked adenosine[J]. J Am Chem Soc, 2004, 126(40): 12720–12721.
DOI:10.1021/ja046964y |
| [12] |
Hwu J R, Jain M L, Tsai F Y, et al. Ceric ammonium nitrate on silica gel for efficient and selective removal of trityl and silyl groups[J]. J Org Chem, 2000, 65(17): 5077–5088.
DOI:10.1021/jo000024o |
| [13] |
Khalafi-Nezhad A, Parhami A, Rad M N S, et al. A catalytic method for chemoselective detritylation of 5'-trity-lated nucleosides under mild and heterogeneous conditi-ons using silica sulfuric acid as a recyclable catalyst[J]. Tetrahedron Lett, 2007, 48(30): 5219–5222.
DOI:10.1016/j.tetlet.2007.05.153 |
| [14] |
Titova E M, Rahaman S M W, Shubina E S, et al. Catalytic redox isomerization of allylic alcohols withrhodium and iridium complexes with ferrocene phos-phine-thioether ligands[J]. J Mol Catal A-Chem, 2017, 426: 376–380.
DOI:10.1016/j.molcata.2016.08.014 |
| [15] |
Ichikawa T, Netsu M, Mizuno M, et al. Development of a unique heterogeneous palladium catalyst for the Suzuki–Miyaura reaction using (hetero) aryl chlorides and chemoselective hydrogenation[J]. Adv Synth Catal, 2017, 359(13): 2269–2279.
DOI:10.1002/adsc.201700156 |
| [16] |
Angle S R, Arnaiz D O. Formal[3+ 2]cycloaddition of benzylic cations with alkenes[J]. J Org Chem, 1992, 57(22): 5937–5947.
DOI:10.1021/jo00048a029 |
| [17] |
Teske J, Plietker B. Fe-Catalyzed cycloisomerization of aryl allenyl ketones: Access to 3-arylidene-indan-1-ones[J]. Org Lett, 2018, 20(8): 2257–2260.
DOI:10.1021/acs.orglett.8b00612 |
| [18] |
Shiina I, Nakata K. The first asymmetric esterification of free carboxylic acids with racemic alcohols using benzoic anhydrides and tetramisole derivatives: An application to the kinetic resolution of secondary benzylic alcohols[J]. Tetrahedron Lett, 2007, 48(47): 8314–8317.
DOI:10.1016/j.tetlet.2007.09.135 |
| [19] |
Gutman A L, Brenner D, Boltanski A. Convenient practical resolution of racemic alkyl-aryl alcohols via enzym-atic acylation with succinic anhydride in organic solvents[J]. Tetrahedron: Asymmetry, 1993, 4(5): 839–844.
DOI:10.1016/S0957-4166(00)80122-6 |
| [20] |
Xie J H, Liu X Y, Xie J B, et al. An additional coordination group leads to extremely efficient chiral iridium catalysts for asymmetric hydrogenation of ketones[J]. Angew Chem Int Ed, 2011, 50(32): 7329–7332.
DOI:10.1002/anie.201102710 |
| [21] |
Chang S J, Zhou S, Gau H M. Enantioselective addition of ArTi (O-i-Pr)3 to aldehydes catalyzed by a titanium complex of an N-sulfonylated amino alcohol[J]. RSC Adv, 2015, 5(13): 9368–9373.
DOI:10.1039/C4RA14173C |
| [22] |
Fernández‐Mateos E, Maciá B, Yus M. Catalytic asym-metric addition of alkyllithium reagents to aromatic aldehydes[J]. Eur J Org Chem, 2012, 2012(20): 3732–3736.
DOI:10.1002/ejoc.201200464 |
| [23] |
Dong J, Du D M. Highly enantioselective synthesis of Warfarin and its analogs catalysed by primary amine-phosphinamide bifunctional catalysts[J]. Org Biomol Chem, 2012, 10(40): 8125–8131.
DOI:10.1039/c2ob26334c |
| [24] |
Wang Y, Zong H, Huang H, et al. Chiral thiophosphoramide catalyzed asymmetric aryl transfer reactions for the synthesis of functional diarylmethanols[J]. Tetrahedron: Asymmetry, 2017, 28(1): 90–97.
DOI:10.1016/j.tetasy.2016.11.011 |
| [25] |
Zhang Zhen-feng(张振锋), Xie Fang(谢芳), Yang Bo(杨波), et al. Chiral phosphoramidite compound and its application in asymmetric catalysis(手性亚磷酰胺配体及其在不对称催化中的应用)[J]. Chin J Org Chem(China)(有机化学), 2016, 31(4): 429-442.
|
| [26] |
Guo Qing-jun(郭庆君). Synthesis of chiral 3-substituted benzoquinone compounds through asymmetric catalytic tandem reaction with chiral phosphoramide as catalyst(手性磷酰胺类化合物不对称催化串联反应合成手性3-取代苯酞化合物)[J]. Chem J Chin Univ-Chin(China)(高等学校化学学报), 2019, 40(10): 1847-1851.
|
| [27] |
Shen B, Huang H, Bian G, et al. Highly efficient asymmetric additions of diethylzinc to aldehydes triply activated by chiral phosphoramide‐Zn (Ⅱ) complexes derived from cinchona alkaloids[J]. Chirality, 2013, 25(9): 561–566.
DOI:10.1002/chir.22171 |