 2024, Vol. 38
2024, Vol. 38



氮杂稠环类化合物是合成多种药物和材料的重要结构单元, 在有机合成化学中得到广泛地应用[1−2]. 其中, 苯并咪唑类化合物在药物、化学和制药工业领域占有非常重要的地位, 被广泛用于糖尿病[3]、抗癌[4−6]、抗病毒和其他疾病的治疗, 是当今医疗行业的研究热点.
苯并咪唑类化合物的合成途径有多种, 其中以邻苯二胺为原料, 和不同的底物进行环合反应是合成苯并咪唑的主要途径之一[7]. Katritzky等[8]以乙酸4-甲基邻苯二胺为原料制得了苯并咪唑衍生物, 但是该方法存在如需要强酸、高温下进行反应条件相对苛刻并且反应周期相对较长; Zhang等[9]以邻苯二胺与原酸酯为原料合成了苯并咪唑类化合物; Das等[10]首次使用了BDMS作为氧化剂氧化芳香醛与邻苯二胺反应, 并且成功合成出了2-芳基苯并咪唑类化合物. 2022年, 卢俊瑞等[11]以邻苯二胺和甲醛为反应模型在无溶剂和无催化剂条件下合成了一系列具有各种生理和药理作用的苯并咪唑类衍生物. 同年, Le课题组[12]以2-硝基苯胺和苯甲醇为底物在元素硫的存在下缩合成2-苯基苯并咪唑. 尽管苯并咪唑类化合物的合成方法目前报道的已有多种, 但是这些方法仍然存在副产物多、毒性大等缺陷[13].
第Ⅳ副族茂金属化合物Cp2TiCl2具有对空气稳定、电子可调等优势, 近年来受到了人们的广泛关注[14−16]. 研究表明: 当二氯二茂钛与不同种类的布朗斯特酸(Brønsted acid)配体协同催化时, 不仅可以提高二氯二茂钛中心金属的Lewis酸性, 还可以提高催化性能及产物的选择性[17]. 我们旨在设计一种双酸催化体系, 用Brønsted acid配体调控二茂钛中心金属钛的Lewis酸性, 从而得到苯并咪唑类化合物.
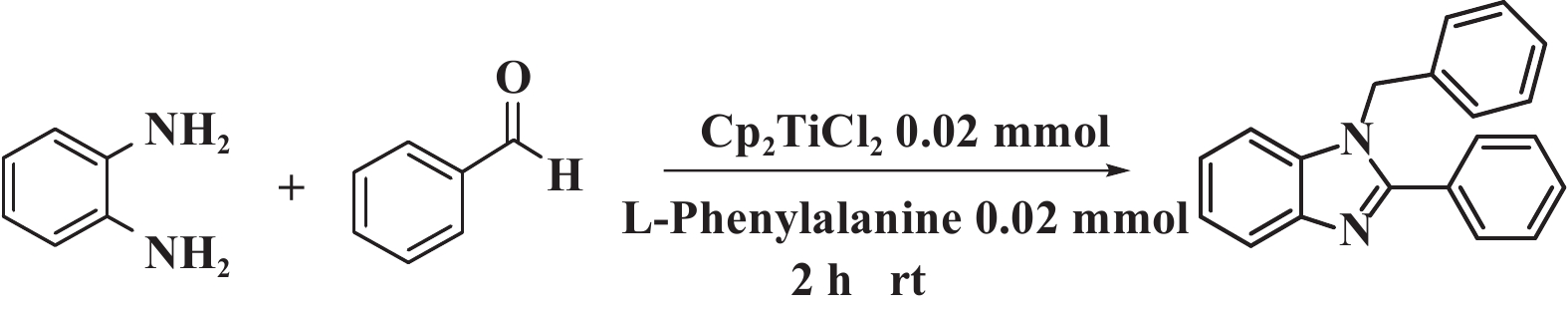
1 实验部分 1.1 仪器与试剂核磁共振仪(ZNCL-TS, 天津市布鲁克科技有限公司); 旋转蒸发仪(RE-2000A, 上海亚荣生化仪器厂); 电子分析天平(ES1020, 上海佑科科学仪器有限公司); 紫外荧光灯(GT61M, 陕西广腾电子科技有限公司) ; 二氯甲烷、三氯甲烷、乙腈、乙醇、甲苯、甲醇、苯等溶剂及药品均为分析纯, 使用前未进一步纯化.
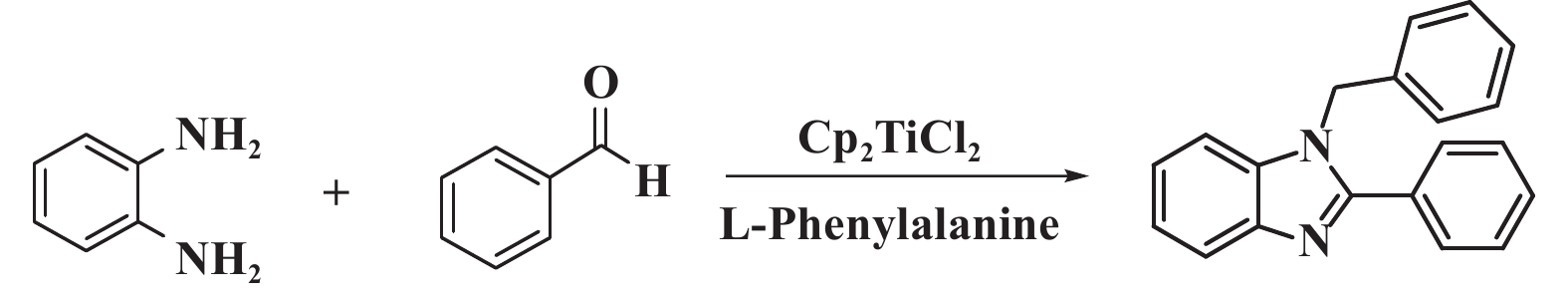
1.2 单/双咪唑类化合物的合成单/双取代苯并咪唑化合物的合成方法如图1所示. 单取代苯并咪唑合成: 向20 mL的反应瓶中加入108.1 mg (1 mmol) 邻苯二胺、122 μL (1.2 mmol)苯甲醛、3.3 mg (0.02 mmol) L-苯丙氨酸、2 mL无水乙醇, 在50 ℃下搅拌8 h, 用硅胶柱分离(洗脱剂为乙酸乙酯与石油醚的体积比为1∶10的混合液) 得到产物; 双取代苯并咪唑合成: 向20 mL的反应瓶中加入108.1 mg (1 mmol) 邻苯二胺、224 μL (2.2 mmol) 苯甲醛、4.9 mg (0.02 mmol) 二氯二茂钛、6.6 mg (0.04 mmol) L-苯丙氨酸、2 mL无水乙醇, 在50 ℃下搅拌8 h, 用硅胶柱分离(洗脱剂为乙酸乙酯与石油醚的体积比为1∶16的混合液)得到产物.

|
图 1 单/双取代苯并咪唑化合物的合成方法 Fig.1 Method for synthesizing 1,2-disubstituted /monosubstituted benzimidazole compounds |

|
1H NMR (400 MHz, DMSO-d6) δ: 7.76 – 7.69 (m, 3H), 7.53 (dd, J = 5.0, 1.6 Hz, 3H), 7.47 (dd, J = 6.2, 2.6 Hz, 1H), 7.31 – 7.20 (m, 5H), 7.00 (d, J = 7.0 Hz, 2H), 5.59 (s, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 153.72, 143.14, 137.41, 136.36, 130.61, 130.31, 129.51, 129.26, 127.95, 126.56, 123.17, 122.69, 119.74, 111.59.

|
1H NMR (400MHz, DMSO-d6) δ: 7.79 (s, 1H), 7.73 (dd, J = 7.3, 2.3 Hz, 2H), 7.53 (dd, J = 7.2, 1.3 Hz, 3H), 7.28 (t, J = 6.7 Hz, 4H), 6.98 (d, J = 6.8 Hz, 2H), 5.60 (s, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 155.22, 143.98, 137.07, 135.22, 130.61, 130.14, 129.54, 129.30, 128.04, 127.19, 126.55, 123.30, 119.18, 113.06, 48.09.

|
1H NMR (400MHz, DMSO-d6) δ: 7.75 (dq, J = 15.1, 5.1 Hz, 3H), 7.52 – 7.48 (m, 1H), 7.44 – 7.18 (m, 5H), 7.11 (t, J = 8.8 Hz, 2H), 7.03 (d, J = 5.5 Hz, 2H), 5.56 (s, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 164.63, 160.65, 143.06, 136.27, 133.52, 131.94, 128.77, 127.12, 122.78, 119.77, 116.48, 116.26, 116.20, 115.99, 111.55.

|
1H NMR (400 MHz, DMSO-d6) δ: 7.67 (d, J = 7.3 Hz, 1H), 7.52 (t, J = 7.8 Hz, 1H), 7.39 (dd, J = 23.2, 7.1 Hz, 2H), 7.19 (d, J = 4.4 Hz, 4H), 7.07 (t, J = 7.4 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.75 (t, J = 7.4 Hz, 1H), 6.58 (d, J = 7.4 Hz, 1H), 5.21 (s, 2H), 3.67 (t, J = 2.9 Hz, 6H).
13C NMR (101 MHz, DMSO-d6) δ: 157.53, 156.83, 152.32, 143.36, 135.68, 132.40, 132.03, 129.17, 127.93, 124.65, 122.66, 122.02, 120.98, 120.00, 119.54, 111.96, 111.34, 111.16.

|
1H NMR (400 MHz, DMSO-d6) δ: 7.70 – 7.67 (m, 3H), 7.44 (dd, J = 6.2, 2.7 Hz, 1H), 7.21 (dd, J = 9.2, 3.1 Hz, 2H), 7.09 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.7 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 5.49 (s, 2H), 3.82 (s, 3H), 3.68 (s, 3H).
13C NMR (101 MHz, DMSO-d6) δ: 160.82, 158.97, 153.62, 143.15, 136.31, 130.98, 129.28, 127.87, 122.83, 122.50, 114.71, 111.47, 55.78, 55.48, 47.38.

|
1H NMR (400 MHz, DMSO-d6) δ: 8.54 (d, J = 2.1 Hz, 1H), 8.17 (dd, J = 8.9, 2.2 Hz, 1H), 7.93 (d, J = 8.9 Hz, 1H), 7.78 (d, J = 7.9 Hz, 2H), 7.56 (d, J = 7.3 Hz, 3H), 7.28 (dt, J = 12.5, 6.9 Hz, 3H), 7.01 (d, J = 7.0 Hz, 2H), 5.77 (s, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 158.79, 147.62, 143.39, 136.79, 135.90, 131.19, 129.66, 129.55, 129.42, 129.38, 126.59, 118.53, 108.64, 48.29.

|
1H NMR (400 MHz, DMSO-d6) δ: 7.75 – 7.64 (m, 5H), 7.48 (d, J = 6.7 Hz, 3H), 7.26 (dd, J = 6.2, 2.5 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 5.57 (s, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 152.59, 143.06, 136.74, 136.33, 132.34, 132.19, 131.46, 128.79, 124.06, 123.50, 122.95, 121.08, 119.88, 111.60, 47.36.

|
1H NMR (400 MHz, DMSO-d6) δ: 7.88 (s, 1H), 7.77 – 7.68 (m, 3H), 7.56 – 7.43 (m, 3H), 7.31 – 7.20 (m, 4H), 6.91 (d, J = 7.7 Hz, 1H), 5.61 (s, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 152.02, 142.96, 140.09, 136.38, 133.17, 132.65, 132.08, 131.51, 131.44, 130.95, 129.61, 128.38, 125.53, 123.69, 123.05, 122.50, 122.43, 111.60, 47.32.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.91 (d, J = 8.0 Hz, 1H), 7.75 (s, 1H), 7.51 (t, J =7.5 Hz, 2H), 7.44 – 7.27 (m, 6H), 7.15 (s, 1H), 6.97 (d, J = 7.2 Hz, 1H), 5.44 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 152.53, 143.06, 138.19, 135.94, 135.22, 135.00, 131.61, 130.54, 130.20, 130.10, 129.53, 128.32, 127.05, 126.21, 124.08, 123.68, 123.15, 120.32, 110.37, 47.91.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.91 (d, J = 7.9 Hz, 1H), 7.45 (d, J = 4.7 Hz, 3H), 7.42 – 7.26 (m, 4H), 7.25 – 7.21 (m, 1H), 7.04 (t, J = 8.4 Hz, 1H), 6.91 (d, J = 7.7 Hz, 1H), 6.84 (d, J = 9.3 Hz, 1H), 5.48 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 163.97, 152.61, 143.05, 138.69, 135.96, 130.94, 130.52, 124.82, 123.61, 123.11, 121.52, 120.31, 117.24, 117.03, 116.55, 116.32, 115.16, 114.95, 112.99, 47.92.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.99 (d, J = 1.7 Hz, 1H), 7.67 (dd, J = 7.8, 1.4 Hz, 2H), 7.53 – 7.44 (m, 3H), 7.38 – 7.30 (m, 4H), 7.11 – 7.03 (m, 3H), 5.44 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 155.21, 135.00, 130.26, 129.26, 129.18, 128.89, 128.00, 126.06, 125.91, 122.83, 115.70, 111.81, 48.53.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.76 (d, J = 8.2 Hz, 1H), 7.68 (dd, J = 7.5, 1.7 Hz, 2H), 7.44 (d, J = 7.0 Hz, 3H), 7.38 – 7.28 (m, 3H), 7.19 – 6.98 (m, 4H), 5.42 (d, J = 2.5 Hz, 2H), 2.47 (d, J = 25.1 Hz, 3H).
13C NMR (101 MHz, Chloroform-d) δ: 153.74, 141.32, 136.60, 136.40, 133.12, 130.23, 129.78, 129.25, 129.20, 129.09, 129.06, 128.74, 127.73, 125.98, 125.92, 124.33, 119.53, 110.33, 48.25, 21.90.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.70 (dd, J = 7.9, 1.5 Hz, 2H), 7.50 – 7.43 (m, 3H), 7.33 (d, J = 1.3 Hz, 4H), 7.17 – 7.06 (m, 4H), 5.45 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 154.90, 136.95, 135.94, 130.21, 129.54, 129.14, 128.76, 127.96, 125.96, 124.79, 123.50, 122.66, 109.29, 48.71.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.93 – 7.87 (m, 1H), 7.59 – 7.55 (m, 2H), 7.53 – 7.46 (m, 3H), 7.38 – 7.26 (m, 8H).
13C NMR (101 MHz, Chloroform-d) δ: 152.42, 143.01, 137.22, 137.00, 129.88, 129.46, 128.57, 128.32, 127.43, 123.01, 110.49, 1.06.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.76 – 7.70 (m, 1H), 7.29 (dt, J = 5.7, 3.3 Hz, 1H), 7.24 – 7.18 (m, 2H), 4.09 – 4.03 (m, 2H), 2.89 (q, J = 7.5 Hz, 2H), 1.84 (dt, J = 14.8, 7.4 Hz, 2H), 1.47 (d, J = 15.1 Hz, 3H), 0.97 (t, J = 7.4 Hz, 3H).
13C NMR (101 MHz, Chloroform-d) δ: 156.02, 142.61, 135.16, 121.89, 121.64, 119.18, 109.25, 45.11, 23.15, 20.81, 11.91, 11.47.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.91 – 7.58 (m, 3H), 7.47 (d, J = 7.4 Hz, 3H), 7.40 – 7.28 (m, 3H), 7.22 – 6.93 (m, 3H), 5.41 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 156.11, 135.57, 135.44, 130.37, 129.29, 129.17, 128.93, 128.14, 125.91, 125.85, 111.44, 106.92, 106.68, 98.47, 98.20, 48.72, 48.67.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.94 (d, J = 8.4 Hz, 1H), 7.69 (d, J = 6.7 Hz, 2H), 7.59 – 7.44 (m, 5H), 7.36 (d, J = 7.3 Hz, 3H), 7.09 (d, J = 6.5 Hz, 2H), 5.51 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 156.61, 145.32, 135.67, 135.56, 130.50, 129.30, 128.95, 128.14, 125.86, 120.41, 119.81, 108.17, 48.58.

|
1H NMR (400 MHz, Chloroform-d) δ: 8.12 (s, 1H), 7.66 (d, J = 6.8 Hz, 2H), 7.51 – 7.44 (m, 4H), 7.36 (d, J = 7.5 Hz, 3H), 7.06 (d, J = 6.6 Hz, 2H), 5.41 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 155.97, 143.55, 136.25, 135.45, 130.51, 129.32, 128.96, 128.18, 125.81, 124.45, 118.06, 114.99, 48.59.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.93 (s, 1H), 7.69 – 7.64 (m, 2H), 7.48 (tt, J = 8.7, 4.7 Hz, 3H), 7.39 – 7.28 (m, 4H), 7.07 (d, J = 6.5 Hz, 2H), 5.42 (s, 2H).

|
1H NMR (400 MHz, Chloroform-d) δ: 7.63 (dd, J = 7.4, 2.2 Hz, 2H), 7.55 (s, 1H), 7.46 – 7.38 (m, 3H), 7.38 – 7.29 (m, 3H), 7.20 – 7.15 (m, 1H), 7.13 (d, J = 6.7 Hz, 2H), 6.59 (s, 1H), 5.41 (s, 2H), 4.07 (s, 2H), 3.73 (s, 3H).
13C NMR (101 MHz, Chloroform-d) δ: 156.82, 153.49, 130.22, 129.68, 129.14, 128.72, 127.78, 125.99, 120.49, 94.29, 55.88, 48.40.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.74 (dd, J = 5.5, 3.3 Hz, 1H), 7.31 – 7.26 (m, 1H), 7.21 (dd, J = 5.8, 3.3 Hz, 2H), 3.93 (d, J = 7.4 Hz, 2H), 2.80 (t, J = 11.3 Hz, 1H), 1.92 (s, 4H), 1.87 (s, 1H), 1.85 (d, J = 3.3 Hz, 1H), 1.79 (s, 1H), 1.73 (d, J = 4.6 Hz, 2H), 1.69 – 1.57 (m, 3H), 1.40 (d, J = 7.6 Hz, 3H), 1.22 (d, J = 29.5 Hz, 4H), 1.12 – 1.00 (m, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 159.46, 142.69, 121.67, 121.55, 119.22, 109.72, 49.76, 38.68, 36.39, 32.16, 31.10, 26.47, 26.21, 25.78.

|
1H NMR (400 MHz, Chloroform-d) δ: 7.90 (d, J = 7.9 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.70 (d, J = 8.2 Hz, 2H), 7.63 (d, J = 7.6 Hz, 2H), 7.57 (d, J = 8.0 Hz, 4H), 7.44 (q, J = 8.0 Hz, 5H), 7.35 (dd, J = 12.7, 5.6 Hz, 3H), 7.28 (s, 1H), 7.21 (d, J = 8.0 Hz, 2H), 5.55 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 153.95, 143.31, 142.67, 140.79, 140.11, 135.42, 129.70, 128.86, 127.87, 127.81, 127.44, 127.14, 127.03, 126.43, 123.14, 122.79, 120.04, 48.27.

|
1H NMR (400 MHz, DMSO-d6) δ: 12.92 (s, 1H), 8.20 (s, 2H), 7.57 (s, 3H), 7.49 (t, J = 7.2 Hz, 2H), 7.21 (s, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 151.68, 144.27, 135.47, 129.42, 126.90, 123.00, 122.13, 119.35, 111.79

|
1H NMR (400 MHz, DMSO-d6) δ: 12.73 (s, 1H), 7.83 (dd, J = 8.0, 1.0 Hz, 1H), 7.77 (dd, J = 7.6, 1.7 Hz, 1H), 7.58 – 7.54 (m, 2H), 7.47 (dd, J = 7.7, 5.9 Hz, 1H), 7.28 – 7.21 (m, 3H).
13C NMR (101 MHz, DMSO-d6) δ: 150.89, 133.87, 132.89, 131.84, 129.42, 128.27, 126.90, 122.02, 111.95.

|
1H NMR (400 MHz, DMSO-d6) δ: 13.03 (s, 1H), 8.38 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 1.6 Hz, 2H), 7.52 (t, J = 7.9 Hz, 2H), 7.26 – 7.21 (m, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 150.08, 132.90, 131.65, 129.35, 125.84, 122.73.

|
1H NMR (400 MHz, DMSO-d6) δ: 12.11 (s, 1H), 8.33 (dd, J = 7.8, 1.7 Hz, 1H), 7.66 – 7.60 (m, 2H), 7.51 – 7.46 (m, 1H), 7.26 – 7.10 (m, 5H), 4.03 (s, 3H).
13C NMR (101 MHz, DMSO-d6) δ: 157.49, 156.79, 143.33, 132.40, 129.17, 127.90, 124.61, 122.67, 111.12, 55.74.

|
1H NMR (400 MHz, DMSO-d6) δ: 13.02 (s, 1H), 8.07 – 8.01 (m, 1H), 7.98 (ddd, J = 10.3, 2.5, 1.6 Hz, 1H), 7.69 (s, 1H), 7.64 – 7.55 (m, 2H), 7.34 (td, J = 8.8, 2.9 Hz, 1H), 7.24 (s, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 164.14, 161.72, 150.41, 133.00, 131.66, 131.58, 122.97, 117.17, 116.96, 113.60, 113.55, 111.59.

|
1H NMR (400 MHz, DMSO-d6) δ: 13.30 (s, 1H), 8.42 (s, 4H), 7.74 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.27 (dt, J = 15.0, 7.4 Hz, 2H).
13C NMR (101 MHz, DMSO-d6) δ: 149.48, 148.32, 136.42, 127.88, 124.75, 124.13, 122.90, 119.91, 112.37.

|
1H NMR (400 MHz, DMSO-d6) δ: 13.25 (s, 1H), 8.19 – 8.16 (m, 2H), 7.85 (s, 1H), 7.60 – 7.53 (m, 4H).
13C NMR (101 MHz, DMSO-d6) δ: 154.30, 129.76, 129.53, 127.21.

|
1H NMR (400 MHz, DMSO-d6) δ: 13.09 (s, 1H), 8.19 – 8.16 (m, 2H), 7.59 – 7.49 (m, 5H), 7.36 – 7.33 (m, 1H).
13C NMR (101 MHz, DMSO-d6) δ: 130.69, 130.13, 129.48, 127.08.

|
1H NMR (400 MHz, DMSO-d6) δ: 13.10 (d, J = 10.9 Hz, 1H), 8.20 – 8.16 (m, 2H), 7.74 – 7.66 (m, 1H), 7.55 (dt, J = 12.8, 7.0 Hz, 4H), 7.24 (q, J = 9.7, 8.4 Hz, 1H).
13C NMR (101 MHz, DMSO-d6) δ: 153.30, 145.22, 136.22, 130.68, 130.17, 129.47, 127.05, 126.56, 123.09, 118.73.

|
1H NMR (400 MHz, DMSO-d6) δ: 13.30 (s, 1H), 8.16 (dd, J = 8.0, 1.6 Hz, 2H), 8.09 – 7.90 (m, 2H), 7.58 – 7.54 (m, 3H).
13C NMR (101 MHz, DMSO-d6) δ: 154.11, 131.10, 129.67, 129.56, 127.24.

|
1H NMR (400 MHz, DMSO-d6) δ: 13.21 (d, J = 15.8 Hz, 1H), 8.16 (d, J = 7.2 Hz, 2H), 7.71 (d, J = 9.9 Hz, 1H), 7.60 – 7.51 (m, 4H).
13C NMR (101 MHz, DMSO-d6) δ: 153.86, 141.05, 132.29, 130.83, 129.94, 129.49, 120.04, 112.52, 106.36, 99.81.

|
1H NMR (400 MHz, DMSO-d6) δ: 12.16 (s, 1H), 7.46 (s, 2H), 7.10 (d, J = 2.8 Hz, 2H), 2.83 (q, J = 7.6 Hz, 3H), 1.32 (t, J = 7.6 Hz, 6H).
13C NMR (101 MHz, DMSO-d6) δ: 156.60, 135.41, 117.75, 115.02, 22.42, 12.67.

|
1H NMR (400 MHz, DMSO-d6) δ: 12.10 (s, 1H), 7.46 (s, 2H), 7.10 (dd, J = 6.0, 3.2 Hz, 2H), 2.84 (tt, J = 11.5, 3.6 Hz, 1H), 2.04 – 1.99 (m, 2H), 1.80 (dt, J = 12.5, 3.3 Hz, 2H), 1.68 (ddd, J = 13.5, 9.4, 3.3 Hz, 1H), 1.59 (td, J = 12.4, 3.1 Hz, 2H), 1.38 (ddd, J = 15.6, 7.9, 3.1 Hz, 2H), 1.27 (ddd, J = 12.2, 8.9, 3.3 Hz, 1H).
13C NMR (101 MHz, DMSO-d6) δ: 159.34, 38.17, 31.71, 26.05.
2 结果与讨论 2.1 1,2-二取代苯并咪唑类化合物的合成首先, 我们选择以邻苯二胺和苯甲醛作为底物来测试不同的Brønsted acid配体与Cp2TiCl2协同催化时对反应的影响(表1) . 在无催化剂存在的情况下反应所得产物产率为47% (Entry 1) , 而仅有0.02 mmol Cp2TiCl2作为催化剂时所得产物产率为68% (Entry 2) , 说明Cp2TiCl2对反应有一定的促进作用. 接下来在筛选不同的Brønsted acid配体与Cp2TiCl2协同催化对反应的影响时, 发现水杨酸系列相比于苯甲酸系列和苯磺酸系列效果更好, 但最高产率也仅为78% (Entry 4). 而在引入几种氨基酸后(Entry 11−16)经测试表明L-苯丙氨酸催化效果最好, 产率可达到94% (Entry 11). 同时对比了D-苯丙氨酸作为配体的催化效果, 产率仅为37%.
| 表 1 不同配体对 Cp2TiCl2催化合成1,2-取代苯并咪唑的影响a Table 1 Effect of different Ligands on the catalytic synthesis of 1,2-disubstituted benzimidazole by Cp2TiCl2a |
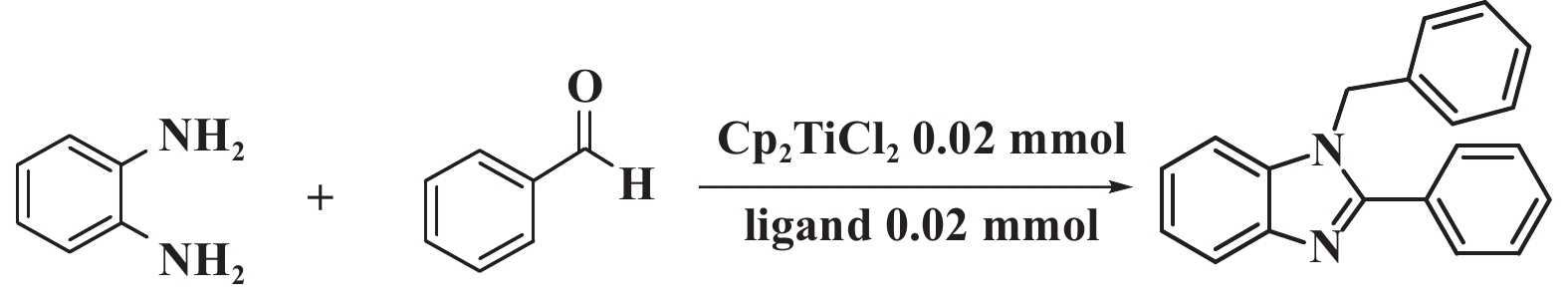
配体筛选表明, 无催化剂或者单独用Cp2TiCl2催化所得产率均不理想, 而以L-苯丙氨酸作为配体与Cp2TiCl2配位在增加中心金属钛路易酸性的同时也提高了反应的产率.
在筛选出最佳配体之后, 我们用单因素实验法对其他反应条件进行了考察. 首先考察了溶剂对反应的影响(表2) . 通过对比无水乙醇、甲醇、苯、甲苯、二氯甲烷、三氯甲烷、乙腈、正己烷等溶剂发现, 该反应在极性较大的溶剂中所得产物产率较高, 如甲醇、无水乙醇, 其中无水乙醇中反应效果最好, 产率达到94%( Entry 1) .
| 表 2 反应溶剂的筛选a Table 2 Screening of reaction solventsa |
在确定了最佳配体和溶剂之后, 紧接着对催化剂用量、比例及反应温度进行了筛选(表3) . 确定二氯二茂钛与L-苯丙氨酸的比例为1∶2时产率较高, 随后在最佳比例不变的基础上加大催化剂用量(Entry 3−6) , 随着催化剂用量的增加, 产物产率略有升高, 最终确定催化剂最佳用量为二氯二茂钛0.02 mmol L-苯丙氨酸0.04 mmol. 最后在最佳溶剂和最佳催化剂用量的基础上探究温度对产物产率的影响, 经对比发现随着温度升高, 产物产率逐渐升高, 温度升高到60 ℃时产率不变. 确定最佳反应温度为50 ℃. 经筛选所得最优反应条件为0.02 mmol二氯二茂钛、0.04 mmol L-苯丙氨酸在2 mL无水乙醇为溶剂下50 ℃反应2 h即可达到97%的产率.
| 表 3 催化剂用量及反应温度的筛选 Table 3 Screening of catalyst dosage and reaction temperature |
接下来, 我们对该催化体系的底物普适性进行了考察. 如图2所示, 连有不同取代基的芳香醛、脂肪醛以及芳香胺在该反应条件下都可以以中等以上产率得到1,2-二取代苯并咪唑类化合物. 首先我们考察了芳香醛上连有不同取代基的影响, 实验研究发现, 连有供电子体基团(3i、3j) 的芳香醛反应所得到的1,2-二取代苯并咪唑类化合物, 其收率分别为64%、86%, 考虑空间位阻的影响, -OMe在邻位时空间位阻相对于对位来说较大, 产率有所下降. 连有吸电子基(3b、3c、3g、3h、3k) 的芳香醛同样可以得到中等以上产率, 而对于活性较低的脂肪醛(3u、3v) 也有一定的收率. 紧接着, 我们又对连有不同基团的芳香胺进行了考察, 发现含给电子基团(3p、3q) 的芳香胺产率为47%和66%; 而含吸电子基团(3d−f、3r−t) 的芳香胺均有中等收率. 结合上述底物普适性考察, 说明不同取代基的电子效应及空间效应对反应的收率有一定的影响, 但差异并不大, 证明了该催化体系具有良好的底物普适性.

|
图 2 合成1,2-二取代苯并咪唑类化合物的底物范围.所有反应均用1.0 mmol的1和2.2 mmol的2; 分离产率均为柱色谱纯化后获得 Fig.2 Substrate scope for the synthesis of 1,2-disubstituted benzimidazole. All reactions were carried out with 1.0 mmol 1 and 2.2 mmol 2. Isolated yields were obtained after purification by column chromatography |
最后, 我们对L-苯丙氨酸与Cp2TiCl2催化合成1,2-二取代苯并咪唑的机理进行了初步的研究. 采用核磁氢谱滴定跟踪二氯二茂钛和配体配位过程中的茂环峰化学位移值的变化(图3) .

|
图 3 2 μmol Cp2TiCl2和4 μmol L-苯丙氨酸的1H NMR滴定实验(新产生的Cp H峰◆, Cp2TiCl2 Cp H峰●) Fig.3 1H NMR titration experiment of 2 μmol Cp2TiCl2 and 4 μmol L-Phenylalanine(newly generated Cp H Peak◆, Cp2TiCl2 Cp H Peak ●) |
首先分别将2 μmol Cp2TiCl2和4 μmol L-苯丙氨酸溶解于0.5 mL氘代甲醇中并分别扫描核磁共振氢谱后发现Cp2TiCl2在δ=6.61处有一茂环特征峰, 而在6.67~6.60之间没有L-苯丙氨酸的质子峰. 为模拟催化剂体系还原实验条件, 将2 μmol Cp2TiCl2和4 μmol L-苯丙氨酸混合在0.5 mL氘代甲醇中, 待全部溶解后, 原位扫描Cp2TiCl2与L-苯丙氨酸的混合谱图(1) , 可以用特征环戊二烯基(Cp) 氢的化学位移值判断新的茂钛络合物的形成. 每隔1 h进行一次核磁扫描, 将扫描7次的核磁氢谱与二氯二茂钛及L-苯丙氨酸的核磁氢谱叠加(图3). 可以观察到在未加入L-苯丙氨酸时Cp2TiCl2的特征茂环峰在δ=6.61处(●) , 加入L-苯丙氨酸后6.61处的茂环峰逐渐减弱, 而在δ=6.67处(◆) 产生了新茂环峰, 并随着时间的推移逐渐增强, 说明Cp2TiCl2在氘代甲醇中与L-苯丙氨酸配位生成了新的茂钛配合物.
基于上述核磁滴定实验我们推测双酸协同催化合成1,2-二取代苯并咪唑合成可能的催化循环机理(图4), 反应开始时Cp2TiCl2与 Ⅰ 配位生成配合物Ⅱ的同时释放两分子的HCl, 邻苯二胺则与一分子苯甲醛结合生成亚胺 Ⅲ, 随后 Ⅲ 中氨基上氮原子进攻配合物 Ⅱ 中Ti―O键形成中间体 Ⅳ并释放出一分子L-苯丙氨酸, 而另一分子苯甲醛与Ⅳ亲核加成反应生成 V, 紧接着中间体V收到体系中释放出的HCl质子化脱水得中间体VI, VI中亚胺氮原子进攻碳VII, VII经氢转移得到目标化合物并与体系中L-苯丙氨酸重新结合得到配合物分子Ⅱ, 进入下一步循环.

|
图 4 可能的催化循环 Fig.4 Possible Catalytic cycle |
在利用薄层色谱法追踪邻苯二胺和苯甲醛合成1,2-二取代苯并咪唑过程中发现, 反应体系仅用L-苯丙氨酸催化时, 在紫外灯下观察仅有一个产物点生成且该点不同于1,2-二取代苯并咪唑产物点, 经柱层析分离并提纯产物后利用NMR确定产物为单取代苯并咪唑, 但产率较低仅有40%. 接下来对单取代苯并咪唑的合成进行了条件优化(表4) .
| 表 4 反应条件的优化 Table 4 Optimization of reaction conditions |
通过筛选反应溶剂(Entry 1−7) , 我们发现该反应在乙醇中仍然可以得到相对较高的收率(Entry 4) . 随后对反应物比例及催化剂用量进行考察(Entry 8−15) , 发现当底物邻苯二胺: 苯甲醛为1∶1.2, 催化剂L-苯丙氨酸用量为0.02 mmol时效果最佳. 最后对反应温度进行考察(Entry 16−18) , 实验表明, 产率随温度升高而增加, 升温至70 ℃时产率不变. 经筛选确定最佳反应条件为50 ℃下以2 mL无水乙醇为溶剂0.02 mmol L-苯丙氨酸催化下1 mmol邻苯二胺与1.2 mmol苯甲醛反应8 h可得到单取代苯并咪唑, 产率可达到70%.
在筛选出最优反应条件以后, 同样我们对底物普适性进行了考察(图5) , 分别测试了不同基团取代芳香醛(3b−3f) 均得到中等及以上产率, 其中邻位溴代的芳香醛(3b) 较间位溴代的芳香醛( 3c) 的产率略有下降, 考虑可能是受空间位阻的影响; 脂肪族醛(3i、3m) 也得到中等收率; 带有不同取代基的芳香胺均有中等以上的收率.

|
图 5 单取代苯并咪唑类化合物的底物范围(反应条件:所有反应均用1.0 mmol的1和1.2 mmol的2; 核磁产率) Fig.5 Range of substrates for the synthesis of monosubstituted benzimidazoles (Reaction conditions: all reactions were carried out with 1.0 mmol of 1 and 1.2 mmol of 2; Yields were obtained by in-situ 1H NMR |
发展了一种配体调控Cp2TiCl2催化合成单、双取代苯并咪唑衍生物的方法. 二氯二茂钛与L-苯丙氨酸协同作用时得到双取代苯并咪唑衍生物, 而单独使用L-苯丙氨酸催化时得到单取代苯并咪唑的方法. 本方法反应条件温和, 催化体系底物普适性良好, 产率较高. 尽管目前尚未得到有力的证据证明催化活性物种与底物的作用模式, 但我们通过1H NMR手段初步对二氯二茂钛与L-苯丙氨酸协同催化过程中产生的新的催化活性物种进行了追踪检测, 并给出了可能的催化机理. 后续工作中我们将对茂金属配合物催化反应的作用机制做进一步深入研究.
| [1] |
Heterocyclic nanographenes and other polycyclic heteroaromatic compounds: Synthetic routes, properties, and applications[J]. Chem Rev, 2017, 117(4): 3479–3716.
DOI:10.1021/acs.chemrev.6b00076 |
| [2] |
Divergent synthesis of densely substituted arenes and pyridines via cyclotrimerization reactions of alkynyl triazenes[J]. J Am Chem Soc, 2019, 141(26): 10372–10383.
DOI:10.1021/jacs.9b04111 |
| [3] |
Novel 4‐heteroaryl‐antipyrines as DPP‐IV inhibitors[J]. Chem Biol Drug Des, 2015, 86(5): 1292–1303.
DOI:10.1111/cbdd.12593 |
| [4] |
Efficient pyrido [1, 2-a] benzimidazole formation from 2-aminopyridines and cyclohexanones under metal-free conditions[J]. Green Chem, 2016, 18(3): 667–671.
DOI:10.1039/C5GC01978H |
| [5] |
Antiproliferative activity of amino substituted benzo [b] thieno [2, 3-b] pyrido [1, 2-a] benzimidazoles explored by 2D and 3D cell culture system[J]. Eur J Med Chem, 2017, 125: 722–735.
DOI:10.1016/j.ejmech.2016.09.084 |
| [6] |
An electrochemical study on the redox chemistry of cyclic benzimidazole derivatives with potent anticancer activity[J]. Electrochim Acta, 2019, 297: 452–462.
DOI:10.1016/j.electacta.2018.11.198 |
| [7] | |
| [8] |
Zhao Dan-dan(赵丹丹). Synthesis and reaction mechanism of benzimidazole and its derivatives(苯并咪唑及其衍生物的合成及反应机理研究)[D]. Nanjing (南京): Nanjing University of Science and Technology(南京理工大学), 2016.
|
| [9] |
An expeditious synthesis of benzimidazole derivatives catalyzed by Lewis acids[J]. Catal Commun, 2007, 8(7): 1126–1131.
DOI:10.1016/j.catcom.2006.10.022 |
| [10] |
Efficient (bromodimethyl) sulfonium bromide mediated synthesis of benzimidazoles[J]. Tetrahedron Lett, 2007, 48(1): 61–64.
DOI:10.1016/j.tetlet.2006.11.018 |
| [11] |
Sun Yue-ying(孙悦滢), Liu Jin-biao (刘金彪), Lu Bo-wei(卢博为), et al. Solvent-free and catalyst-free synthesis of benzimidazole (无溶剂无催化合成苯并咪唑类衍生物)[J]. J Tianjin Univ Technol (天津理工大学学报), 2021, 37 (05): 61−64.
|
| [12] |
Step-and atom-economical synthesis of 2-aryl benzimidazoles via the sulfur- mediated redox condensation between o-nitroanilines and aryl methanols[J]. Tetrahedron, 2022, 121: 132918.
DOI:10.1016/j.tet.2022.132918 |
| [13] | |
| [14] |
Salicylato titanocene complexes as cooperative organometallic Lewis acid and Brønsted acid catalysts for three-component Mannich reactions[J]. Chem Eur J, 2014, 20(28): 8530–8535.
DOI:10.1002/chem.201402438 |
| [15] |
Solvent strategy for unleashing the Lewis acidity of titanocene dichloride for rapid Mannich reactions[J]. RSC Adv, 2016, 6(19): 15298–15303.
DOI:10.1039/C5RA27094D |
| [16] |
Titanocene dichloride and poly (o-aminophenol) as a new heterogeneous cooperative catalysis system for three-component Mannich reaction[J]. Catal Sci Technol, 2015, 5(9): 4346–4349.
DOI:10.1039/C5CY00793C |
| [17] |
Tunable titanocene Lewis acid catalysts for selective Friedel–Crafts reaction of indoles and N‐sulfonylaldimines[J]. Eur J Org Chem, 2016, 2016(3): 502–507.
DOI:10.1002/ejoc.201501122 |